
Review of EPO Antibody Decisions in 2021
Last year we provided a review of 21 EPO Board of Appeal cases relating to “antibody inventions” that were decided and published between January 2018 and January 2021. At the time, we observed that this relatively small number of decisions was consistent with our view that there had been relatively little case law in this field, despite the high number of applications and patents relating to antibodies that have been prosecuted at the EPO in the same period. Now considering the activity of the Boards of Appeal in the calendar year of 2021, we see some evidence that the trend of minimal case law may at last be reversing. We counted 17 “antibody invention” cases that were decided and published between January and December 2021. Of course, this apparent increase in Board activity in the antibody field may simply reflect an increase in activity generally. The advent of routine video conference Oral Proceedings is likely a factor here. Our experience is that hearing dates are now less likely to be rearranged, whether due to the ongoing coronavirus pandemic or other reasons, and the Boards remain generally keen to clear their backlog of cases. That said, all but 2 of the 17 decisions covered in this review arise from an application with a filing date between 1999 and 2009, which may suggest that even with the increase in activity, a backlog of older cases remains. It is also important to remember that there can be a lag of several months between a case being decided at oral proceedings and the written decision being published. As such there will be cases with a decision date in 2021 that are not covered by this review because they will not publish until 2022.
We do not believe that any of the 17 decisions covered in this review represents a significant shift in EPO practice, or that any of them will compel changes in the way examination is conducted. Instead, we see that the EPO has continued to treat antibody inventions in a manner broadly comparable to other forms of technology. In decisions where “antibody-specific” considerations may be applied, the EPO has done this consistently and in line with the (now largely established) basic principles that are further explored in our advanced guide to drafting and prosecuting antibody inventions at the EPO. The majority of the decisions support the current practices of the examining divisions that we observe in routine prosecution, and which were partly codified in the EPO’s Guidelines for Examination in March 2021 (also discussed in our advanced guide). On a day to day basis the Guidelines are therefore probably more significant than the case law, but the 17 decisions covered in this review nonetheless provide confirmation and consolidation of EPO practice, represent useful examples of fact patterns for future reference, and may also help to illustrate developing trends. Such trends may include the general increase in Board activity in this field, but we also observe an increase in the proportion of cases relating to “downstream” developments of existing antibodies. In addition we see some evidence of a trend towards more antibody inventions seeking to define an antibody “sub class” to provide some breadth around one or more specific antibodies. Such cases may define a sub-class by comparison to a specific antibody, using terms such as “competes for binding”. We expect that this claim type in particular will be the subject of future Boards of Appeal decisions. We have observed some inconsistency in the EPO’s interpretation of such claims during prosecution, and the Guidelines are regrettably silent on this point.
Below we give some statistics and discuss the 17 decisions in more detail.
Statistics
We identified 31 decisions decided and published between January 2021 and December 2021 which refer to the term “antibody”. 14 of these decisions were not concerned with antibody inventions as such and so are not considered further in this view. Of the remaining 17, there was an approximately equal split between appeals from opposition division decisions (8 decisions) and appeals from examining division decisions (9 decisions). Board 3.3.04 unsurprisingly retained its position as the most prolific “antibody Board” with 12 of the 17 decisions (previously 15/21 from 2018 – 2021). 4 decisions were issued by Board 3.3.08 (previously 1/21 from 2018 – 2021), with the final 1 decision issued by Board 3.3.07 (previously 2/21 from 2018 – 2021). We identified no antibody decision by Board 3.3.01 (previously 3/21 from 2018 – 2021), which likely reflects the absence in this review of decisions relating to “clinical formulations” of antibodies. We discerned no obvious differences in practice between the various Boards.
A breakdown of the key grounds for each decision is provided in Figure 1.
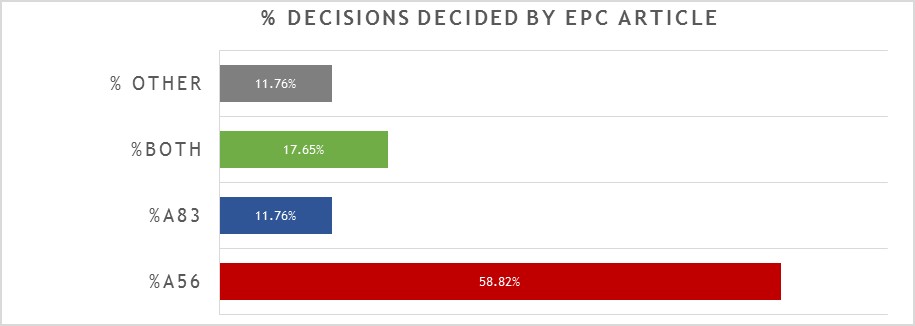
Repeating the trend from previous years, close to 60% of the 17 decisions were decided primarily on the basis of inventive step (Article 56 EPC), with a slightly reduced proportion (around 12%) decided primarily on the basis of sufficiency (Article 83 EPC), and around 18% considering both inventive step and sufficiency to some degree. It remains the case that there is often considerable overlap between Article 56 EPC and Article 83 EPC for antibody inventions, and we found the EPO’s assessment of both Articles to be consistent across all of the relevant decisions. Outside of Article 56 EPC and Article 83 EPC, only 2 decisions were decided on another ground. In one case (see T2898/18) the ground was Article 123(2) EPC: correction of an error in the ATCC reference number for a deposited hybridoma expressing an antibody was held to add matter, fatally undermining the entire case. In the other case (see T2248/18) the ground was Article 84 EPC: an internal designation “antibody ACZ885” was held to be unclear and the patentee was refused permission (on procedural grounds) to enter a clarifying amendment. Therefore, whilst both cases were decided by formal objections to the features mentioned, it can be argued that the underlying problem was again sufficiency under Article 83 EPC: the skilled person cannot carry out the invention if the relevant features are not correct / clear. There are no other, new “antibody-specific” issues illustrated by these two cases. However, since internal designations and deposited hybridomas are relatively common components in antibody inventions, they may nonetheless represent cautionary tales for applicants in the field.
The 17 decisions can also be divided into four general categories of invention as shown in Figure 2.
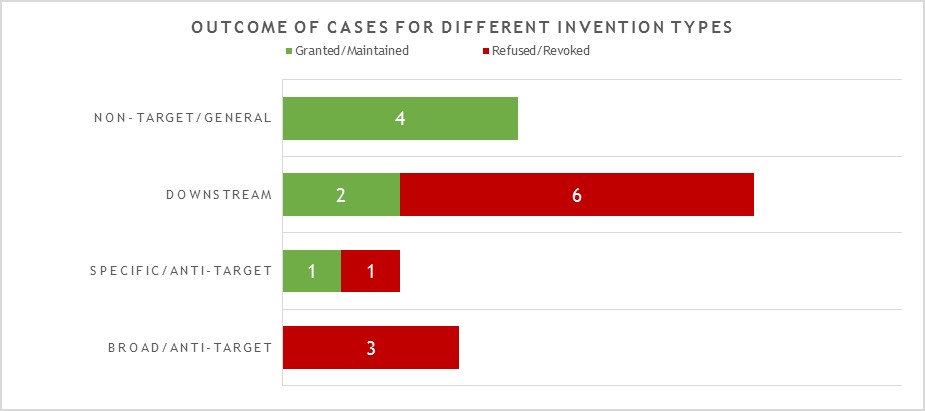
This year the largest category (8 decisions – 2 granted/maintained, 6 revoked/refused) is concerned with what we define as “downstream” antibody inventions. These inventions relate to advances which typically arise during downstream development of a pharmaceutical, such as combinations with other therapeutic agents, or identification of alternative medical uses, sub groups of patients, dosing regimens, partner diagnostics, and the like. In many respects these are not specifically “antibody inventions” because the target binding properties of the antibodies concerned have usually already been established in the art. We find that such inventions are examined by the EPO in the same way as comparable inventions for non-antibody pharmaceuticals. We identified no particular trends in the spread of positive and negative decisions for the applicant/patentee in this category in 2021. However, it is interesting to see that there are proportionally more decisions in this category than in previous years. We expect this trend to continue.
The next largest category (4 decisions – all granted/maintained) is concerned with antibody inventions that we define as “non-target” or “general” in nature, and is similar to the above in that inventions in this category are not typically subject to any “antibody-specific” considerations. Rather they are examined in broadly the same way as any other technology at the EPO. Also similar to the above, we do not see any particular trend in the fact that all four decisions happened to be positive for the applicant/patentee in 2021. It is though pleasing to see that general platforms and methods of manufacture of antibody remain patentable at the EPO.
The next category (3 decisions – all revoked/refused) is concerned with what we define as “broad” anti-target antibody inventions. The earliest form of application/patent in this category is sometimes described as a “1st generation” antibody case. Such cases may entirely lack disclosure of any individual antibody molecule that binds to the target, especially if the invention relates to the identification of the target and/or a link to a particular indication, rather than to an individual antibody or antibodies. The backlog at the Boards of Appeal means that there are likely still some cases of this type to be decided, but we did not observe any this year. Rather, the three cases in this category in 2021 represent variations of a narrower, more focused approach. Rather than seeking to cover a very broad concept, such as all antibodies to a target and/or for an indication, these cases may include examples of specific antibodies but seek to protect all members of a “sub-class” of anti-target antibody defined around those examples. The “sub-class” may relatively broad or relatively narrow depending on the case, and may be defined in the claims with functional and/or structural features. There is often also an explicit feature in the claims that makes comparison to a specific reference antibody. In 2021, the broadest version (presented in T2423/18) of this approach sought to claim a sub-class of anti-target antibody with only a single functional feature (binding specificity for multiple allelic forms). This was held to be a known/obvious feature from prior art antibodies. The narrowest version (presented in T0941/16) defined a sub-class with limited scope for structural variation relative to a reference antibody (replacement of one or more of the 6 CDRs) provided certain functional characteristics were retained. Such inventions are (arguably) only a little broader than the “specific/2nd generation” antibody inventions discussed in the next category. In this case, though, the patent was still revoked as insufficient across the scope sought. The final, intermediate version of the “sub class” approach (presented in T2898/18) is perhaps the most interesting. The antibody “sub class” was here defined by direct comparison to a specific antibody as a reference, with which members of the sub-class “compete for binding” to the target. We have observed some inconsistency in the EPO’s interpretation of claims containing technical features of this type. That being the case, it is disappointing that the Board were able to dispose of this particular case on formal grounds without substantive consideration of the technical features. We expect the “sub-class” type of antibody invention to be the subject of more decisions of the Boards of Appeal in future.
The final category (2 decisions – 1 granted/maintained, 1 revoked/refused) is concerned with what we define as “specific” or “2nd generation” antibody inventions. Typically the invention relates to an individual antibody or antibodies, defined by sequence, and directed to a well-characterised target for which prior art antibodies (often therapeutic antibodies) are known. Readers are likely familiar with the EPO’s approach to such inventions. All antibodies specific for a target are obvious by comparison to prior art antibodies for the same target, unless an unexpected effect is plausibly demonstrated. This can be particularly challenging when the prior art includes high-affinity therapeutic antibodies to the same target. In this context, it is not surprising that both decisions in this category featured Article 56 EPC (inventive step) as a key ground. Both cases essentially relied upon successful treatment of a specific, new indication as the unexpected effect. In one case this was held to be inventive, in the other it was not, but the assessment approach used by the EPO was consistent for both. It is perhaps a little surprising that there are not more decisions in this category, given that we find this to be the most common type of antibody invention currently under prosecution at the EPO. However, it may be that the relatively narrow scope of these “specific” claims is less likely to be subject to opposition.
Broad / Sub-class Decisions
T 2423/18 (MICA binding agents/INNATE PHARMA) concerned any antibody capable of binding to four specific allelic variants of MICA, for treating cancer. Multi-allelic binding was held to be known from mouse antibodies in the art, and it was obvious to adapt these for treatment of cancer in humans. The claim did not require treatment efficacy in all forms of cancer expressing the allelic variants.
T 0941/16 (anti-PSMA antibody/UNIVERSITÄTSKLINIKUM FREIBURG) concerned any anti-PSMA antibody having at least 3 of 6 specific CDRs, plus certain functional characteristics. The applicant argued that starting from the known CDRs the skilled person was able to make any/all humanised variants with any 3 of the 6 and the required functional features. Supporting evidence was filed. The Board found that this evidence, if anything, rather proved that even insisting on what are typically regarded as the two most important CDRs (the third CDR of both the heavy chain and the light chain) did not reliably achieve the desired characteristics, and so the skilled person is not in a position reliably to obtain substantially all antibodies / fragments within the scope of the claims. Thus there is an undue burden under Article 83 EPC (insufficiency). Various elements of the functional features (e.g. references to “minimal” and “strong” binding) were also unclear without further definition. Separately, the patentee had already secured a granted patent to claims defining an antibody by all 6 CDRs.
T 2898/18 (Antibody binding to alpha-synuclein/PROTHENA) concerned an antibody defined as competing for binding to target with a reference antibody and also as binding to an epitope within a specific 20 amino acid region of the target. The Opposition Division were satisfied that the addition of the requirement for the epitope being within a specific region was enough to render it plausible that all antibodies within the scope of the claim would have treatment effects comparable to the reference antibody, and that this treatment effect was inventive over the prior art. However, sufficiency/inventive step was however not considered by the Board because they instead disposed of the case for added matter under Article 123(2) EPC. The added matter arose in the definition of the reference antibody, which included recital of the ATCC deposit number of the hybridoma capable of expressing it. The application as filed had included an error in the ATCC number where it was explicitly identified as expressing the reference antibody, and where the ATCC deposit number was recited correctly elsewhere in the application, it was not clear that it expressed the reference antibody. Thus, the recital in the claims of the correct ATCC deposit number as expressing the reference antibody was held to add matter under Article 123(2) EPC. Attempts to refer to external documents (e.g. searches of ATCC catalogue etc) were held not to be part of the application as filed and thus could not cure the problem. The patentee’s case was not helped by their relatively late recognition of the severity of the problem. In particular, attempts to correct the ATCC deposit number as an obvious error were not considered as they were deemed to be filed late.
Specific/2nd Generation Decisions
T 0096/20 (Treatment of myasthenia gravis/ALEXION) concerned any anti-C5 antibody inhibitor of complement C5 for treating myasthenia gravis (MG). The patentee limited during proceedings to focus on a specific antibody by name (eculizumab). The Board held it to be obvious to use anti-C5 antibody inhibitor of complement C5 to treat MG, with a reasonable expectation of success given a published announcement / protocol for a phase II safety/efficacy trial of eculizamb for MG. There was no change to this expectation just because there had been no new treatment for MG for a long time, nor that trials for other complement directed therapies for other diseases had failed. This illustrates that clinical trial protocol publications can often be extremely difficult prior art to overcome, similar to the situation for non-antibody pharmaceuticals.
T 0033/19 (Eculizumab for use in treating aHUS/ALEXION) concerned the same antibody (eculizumab) as above. The examining division had rejected the case under Article 83 EPC because the application does not demonstrate experimentally that treatment of aHUS (Atypical Hemolytic Uremic Syndrome) is achieved. However, Board reversed this decision on the basis that the application does identify aHUS as complement associated and explicitly suggests treatment with eculizumab, referring to the amelioration of some symptoms. The Board also found that specific documents considered to be representative of common general knowledge (CGK) showed that the etiology of aHUS is known to be complement-mediated and that use of anti-C5 antibodies was thus a potential treatment option. The Board indicated that there is no requirement for direct experimental proof, provided skilled person has no reason to doubt that the agent achieves the claimed effect, with due regard to CGK. The Board went on to review Article 56 EPC (inventive step) over the same CGK documents. The suggestions in the CGK relied upon for Article 83 EPC (i.e. to use anti-C5 for aHUS) were held not enough to provide reasonable expectation of success, whereas the application included an explicit statement that eculizumab has been found to ameliorate some aHUS symptoms. The Board found there was no reason to doubt this statement (despite lack of data in the application), and also that subsequently published data was supportive of the statement. Thus, the Board concluded there is an inventive contribution over these specific CGK documents. The case was remitted to the examining division to consider patentability over other cited documents.
Downstream Decisions
T 0208/17 (Prognostic method for panitumumab/AMGEN) concerned a partner diagnostic for panitumumab, i.e. a method of predicting responsiveness of colorectal adenocarcinoma to the antibody based on the presence or absence of a K-ras mutation. Nothing in the decision is specific to antibodies, but may be of interest in that it shows that downstream antibody-related inventions may be pursued in the same way as for any other pharma product.
T 0795/17 (Antibody 8H9 for treatment of brain cancer/SLOAN KETTERING) concerned a specific antibody defined by 6 CDRs with a conjugated radioisotope, plus specific dose and administration route (intrathecal), for treating primary brain cancer / metastatic CNS cancer. The antibody was known to bind brain tumour tissue, and had previously been used via the intravenous route to treat non-brain cancers when conjugated to a radioisotope. The Board defined the problem as essentially the alternative therapeutic use (in brain) of a known compound. Patentee tried to argue that general difficulties relating to the treatment of brain made application of the compound to this area non-obvious, and presented some examples of other anti-cancer antibodies which had worked in non-brain tissues but not in brain. The Board was not convinced. Documents on file (including the closest prior art) showed that other antibody-isotope conjugates had been used to treat CNS cancers, so the skilled person would not have been discouraged from the attempt by a sub-set that had failed. Intrathecal administration was seen as an obvious route for treating brain / CNS, and the choice of a specific dose was viewed as routine optimisation. The patent was revoked for lack of inventive step.
T 2167/17 (Combination therapy/GENENTECH) concerned a combination of anti-CD20 (effectively rituximab) plus a BLyS antagonist for depleting B cells in a human in need thereof. The BLyS antagonist could also be an antibody. It was found to be obvious from the art that B cell depletion by anti-CD20 was likely to be enhanced by also blocking the BLyS pathway. An attempt to argue that depletion specifically of Marginal Zone and Germinal Centre B cells was an additional effect (in the context of a claim request limited to those cell types) was not considered, being deemed a late change to the patentee’s case. The combination was treated in broadly the same way as any other invention relating to a combination of pharmaceuticals. See also T2168/18 below, between the same parties.
T 2168/17 (Combination therapy 2/GENENTECH) concerned a combination of anti-CD20 (effectively rituximab) plus a BLyS antagonist for treating autoimmune disease. Treating both pathways in the context of autoimmune disease was considered to be obvious over the prior art. As with T2167/17 above, the combination was treated in broadly the same way as any other invention relating to a combination of pharmaceuticals.
T 0707/18 (Campath-1H/ALCAFLEU) concerned a Swiss-style claim to Campath (alemtuzumab) for MS, with a specific patient definition (relapsing, prior treated) and dose regimen. It was not disputed that Campath is a known treatment for MS, but the Examining Division had rejected the claim under Article 83 EPC (sufficiency) because they considered there to be a lack of evidence that there was a therapeutic benefit for the combination of the specific patient group and dose. The Board overruled this decision – a therapeutic benefit was plausible given the evidence available in the patent and the lack of serious doubts provided by other disclosures. The case was remitted to the Examining Division for consideration of other grounds including Article 56 EPC.
T 2248/18 (TRAPS therapy/NOVARTIS) concerned a claim in which an antibody was described using the internal designation “ACZ885”. This was considered to lack clarity under Article 84 EPC – the skilled person could not be sure precisely which antibody was intended. The patentee argued that 2 sequences in the application were associated with the internal designation and we enough to make the term clear, especially given a reference to a separate published PCT which included the same designation and sequences. The Board held that this did not help because the sequences were clearly too short to be the full chains of an antibody, and hence at least parts of whatever is intended by “ACZ885” would remain undefined and unclear. The applicant’s case was not helped by the fact that they did not realise the problem affected all of their pending requests in the appeal. The Board denied permission to enter a late request to address the problem by amendment.
T 2043/17 (ADCC optimisée/LFB) concerned the selection of a specific patient group that derived a particular therapeutic benefit from treatment with a known antibody. This was found to be obvious over the prior art for reasons consistent with usual EPO practice relating to patient groups in other pharmaceuticals.
T 2044/17 (PCSK9 E670G mutants/KYMAB) concerned an attempt to define a sub-group of patients for treatment with anti-PCSK9 (alirocumab) based on presence in the patient of a specific PCKS9 variant and a particular heavy chain allotype. No evidence of improved treatment in the sub-group was provided. It was held to be obvious to treat the sub-group because (i) the variant was physiologically active, so reasonable to expect alirocumab to bind it (unlike other inactive variants which alirocumab does not bind) and (ii) it is obvious to match the allotype of a therapeutic antibody in order to minimise anti-drug immune responses in the patient. Again this is broadly consistent with EPO practice relating to patient groups in other pharmaceuticals.
Non-target/General Decisions
T 2697/16 (Antibody production/CHUGAI SEIYAKU KABUSHIKI KAISHA) concerned a method of IgG1 antibody production, essentially requiring insertion in a cell of exogenous nucleic acid encoding heavy chain (HC) : light chain (LC) in a ratio of 1:2 or higher (excess copy number of LC). This leads to an improved yield relative to vectors with 1:1 ratio. Problem was not defined as an improved method, because the claims do not require that the HC and LC be controlled by the same promoter (as is the case in the Examples showing improvement) and so in principle the benefit of higher LC copy number could be reversed if it had a poor promoter. Nonetheless, even defined as only an alternative the Board found the method to be non-obvious. Although the ratio of HC:LC polypeptide was known to be important, the Board found that it was not taught in the prior art to alter the copy number of the LC nucleic acid vs the HC.
T 2517/19 (Site-specific conjugation of drugs to antibodies / BYONDIS) concerned an antibody-drug conjugate (ADC), with a linker and drug (a duocarmycin derivative) site-specifically conjugated to any IgG1 antibody through an engineered cysteine at Heavy Chain position 41 (Kabat numbering). Duocarmycin derivatives were known for use in ADC, but Board found that the prior art does not suggest that the claimed conjugation at position 41 is advantageous in terms of reduced hydrophobicity of the resulting ADC. The closest prior art did not identify hydrophobicity as a problem.
T 0159/17 (Chimeric antibodies/ABLEXIS) is primarily concerned with a specific mouse / mouse cell and methods of making chimeric antibodies by expression from the mouse/cell. The expressed chimeric antibody has a particular arrangement of human VH1, human CH1, human upper hinge, and mouse CH2 and CH3. Interestingly the chimeric antibody is claimed independently of the mouse/cell/method and was also found inventive and sufficient in its own right.
T 0578/17 (Antibodies with reduced core fucosylation/SEATTLE GENETICS) concerned a general method of making antibodies with reduced core fucosylation, by culturing a host cell expressing the antibody in a medium with particular types of fucose analogue. This was found to be inventive over the cited prior art, which taught alternative approaches to reducing core fucosylation.
Trends and Conclusions
A majority of the decisions discussed above (12/17) are concerned with inventions that are not directly concerned with the “target binding” properties of antibodies. That is, the inventions are either applicable to antibodies generally, or are downstream developments of a particular pre-existing antibody, the target-binding properties of which have already been established. This makes it more difficult to draw any particular conclusion from these decisions that could reasonably be described as “antibody-specific”. On the other hand, it is reassuring to see that the Boards have approached all of these decisions in a manner which is consistent with the EPO’s treatment of other forms of technology, in particular other pharmaceuticals. We expect more decisions of this type in the future, particularly in the downstream category. Whereas competitors developing their own alternative anti-target antibody have tended not to oppose another company’s (relatively narrow) downstream patents for a specific antibody, biosimilar challengers may increasingly seek to oppose such patents, particularly as any compound per se protection begins to expire. We expect that these “biosimilar oppositions” will increasingly be represented at the appeal stage in the coming years.
A corresponding lack of interest in opposing the (typically earlier) compound per se patents in the specific/2nd generation anti-target antibody category may explain why there were only 2 decisions in this category in the present review (both appeals from examining division decisions), despite this being the most common type of claim that we observe in prosecution. However, even with a relatively small number of such decisions, it is reassuring that the Boards remained consistent in their application of the basic principles of antibody examination as set out in the EPO Guidelines.
We believe a further, interesting trend may be developing for broader anti-target antibody inventions. As discussed above, none of the 3 decisions we reviewed in this category concerned a classical “1st generation” antibody invention with claims to a broad concept, such as all antibodies to a target and/or for an indication. Although we do not think the Boards have yet seen the last of such “1st generation” inventions, especially where the claims concern medical uses of structurally undefined antibodies as opposed to such antibodies per se, it is striking that all 3 of the reviewed decisions instead represent variations of a narrower, more focused approach. That is, the 3 decisions are concerned with claims directed to a “sub-class” of anti-target antibody, defined in a variety of different ways, with the apparent aim of securing some additional breadth around a specific antibody or group of antibodies. We believe this may arise from the manner in which applicants have now adapted to the EPO’s approach for specific/2nd generation anti-target antibody inventions. In particular, many applicants may now pursue to grant a relatively narrow set of claims for a lead antibody (of most commercial interest and/or for which the data supporting an unexpected effect is most readily available), but may frequently also file one or more divisionals to attempt broader protection for a “sub-class” around this lead molecule. Broader claims are of course more likely to trouble both examiners and third parties, and as such may be more likely to reach the Boards of Appeal in one form or another.
We therefore expect decisions relating to such antibody “sub-class” inventions to become more common in the future, continuing the trend observed in this review. We also expect to see a sub-trend within this wider trend. Specifically, we expect the Boards will be asked to consider more claims which define a sub-class as “competing for binding” with a reference antibody. As noted above, we have observed some inconsistency in the EPO’s interpretation of this claim type during prosecution, which may ultimately require resolution by the Boards of Appeal.