
Data Exclusivity and Market Protection in the EU / EEA and UK
In order to place a new medicinal product on the market in Europe, it is necessary first to obtain a marketing authorisation. Most applications for marketing authorisations are made through a centralised procedure with the European Medicines Agency (EMA) which effectively leads to authorisation in all EU/EEA member states1, although other routes are available. Regardless of the route, an application for marketing authorisation must be supported by a dossier, referred to at the EMA as the common technical document (CTD), which demonstrates the quality, safety and efficacy of the medicinal product. The dossier will include non-clinical pharmacology and toxicology data, as well detailed clinical trial reports. Obtaining all of the data required for a new dossier represents a considerable investment of time and resources.
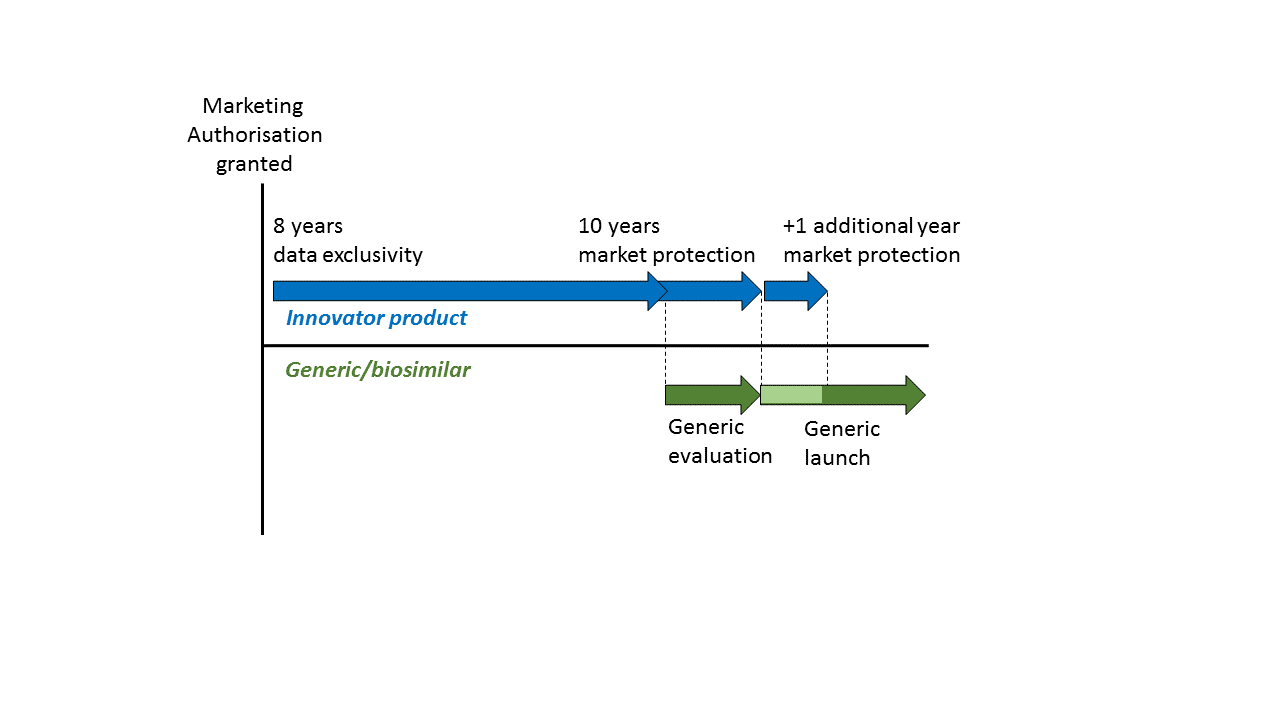
In contrast, an application for marketing authorisation for a generic or biosimilar product does not require the same level of supporting data. It may rely (at least in part) on the published data for the original product. If this were possible immediately after publication, it would significantly undermine the investment in the original product. Accordingly, EU law2 provides for two forms of regulatory data protection that are intended to compensate for this investment. These are referred to as “data exclusivity” and “market protection” and they are defined in Article 14(11) of Regulation (EC) No 726/2004. Regulatory data protection in the EU/EEA (and UK) follows an ‘8+2+1 formula’, providing a maximum of 11 years exclusivity3.
Data exclusivity
A period of 8 years from the initial authorisation of a medicine, during which the marketing-authorisation holder has exclusive rights to the preclinical tests and clinical trials data on the medicine, and during which other applicants cannot rely on these data to support their own applications for marketing authorisation for e.g. generic or biosimilar products.
Market protection
A 10-year period after the initial marketing authorisation of a medicine during which a generic or biosimilar cannot be placed on the market, even if it has already received a marketing authorisation.
The two periods run in parallel, as can be seen from the following representation4:
Data exclusivity and market protection: The 8 + 2 formula
Data exclusivity means that, during a period of 8 years from the initial marketing authorisation of a new medicinal product, the pre-clinical and clinical data contained in the supporting dossier cannot be relied on by other applicants or authorities in subsequent applications to determine the safety and efficacy of other products. Thus, an applicant for a generic or biosimilar product must wait for 8 years if they wish to rely upon this data.
After the 8 year period of data exclusivity has passed, an application for marketing authorisation for a generic or similar may be filed relying upon the data in the original dossier, and may even be granted. However, the generic or similar product may not be placed onto the market until a further 2 years have passed, because of the 10 year period of market protection that also applies from the initial marketing authorisation.
It should be noted that the data exclusivity and market protection provisions do not prevent an application for a marketing authorisation for a generic or similar product that relies instead upon independently generated pre-clinical and clinical data. However, in most cases, the cost of doing so outweighs the benefit of market access before the 8 + 2 years has passed.
Extension of the market protection period: The 8 + 2 (+ 1) formula
The 10 year market protection period may be extended by 1 year if, during the first 8 years, the marketing authorisation holder obtains an authorisation of the medicinal product for a “new therapeutic indication”, where it is considered to provide a “significant clinical benefit” as compared to the other therapies already available for that new indication5.
According to guidance from the European Commission, whilst a “new therapeutic indication” clearly encompasses identifying a new target disease, it can also include identifying the efficacy of the medical product in treating a new stage or severity of a disease, extending the target population group, moving from a combination therapy to monotherapy, or changing from treatment to prevention or diagnosis of a disease.
The significant clinical benefit must be shown in comparison to the already existing therapies for that new therapeutic indication. This includes medicinal products authorised in one or more member states, any non-pharmacological approaches to treating that disease, and any other prior art therapeutic methods for the indication. However, it does not include ‘off-label’ use of existing medicinal products.
In order to demonstrate a “significant clinical benefit” as compared to these existing therapies, the results of comparative clinical studies are generally required. The new treatment must be shown to provide a clinically-relevant advantage or a major contribution to patient care. The European Commission guidance indicates that demonstrating greater efficacy, an improved safety profile, and/or more favourable pharmacokinetic properties may be indicative of a significant clinical benefit. The Committee for Medicinal Products for Human Use (CHMP) or a corresponding National Competent Authority will review applications for significant clinical benefit on a case-by-case basis.
Limits to regulatory data protection – the “Global Marketing Authorisation” (GMA)
When a new medicinal product has been granted an initial marketing authorisation, further authorisations for additional strengths, pharmaceutical forms, administration routes, presentations, or therapeutic indications are considered to belong to the same ‘Global Marketing Authorisation’ (GMA). This includes authorisations granted through separate procedures and under a different name to the holder of the initial authorisation. New authorisations falling within a GMA are not entitled to new periods of data exclusivity and market protection6.
The scope of the GMA has been explained by the European Commission in its “Notice to Applicants” guidance and confirmed by case law from the General Court7. For example, authorisations for different therapeutic indications and different strengths are considered to fall within the GMA. In contrast, combinations of already authorised monotherapies are not considered to fall within the scope of the GMA, and vice versa, assuming that each of the substances in the combination therapy make a therapeutic contribution. In such cases, the new combination or monotherapy would be entitled to a new period of data and market exclusivity.
Summary
Data exclusivity and market protection provide an important layer of protection for new medicinal products, which is complementary to that provided by other rights, such as patents and supplementary protection certificates (SPCs). They may be particularly important where a marketing authorisation is granted relatively close to the expiry of other rights. In these cases, the data exclusivity and market protection periods may be the last rights to expire.
Footnotes
- Post-Brexit, on 1 January 2021 all existing centrally authorised EU MAs were automatically converted into UK MAs effective in Great Britain (England, Scotland and Wales) only (UKMA(GB)). For a period of two years from 1 January 2021, the UK medicines agency (MHRA) may rely on a decision taken by the European Commission (EC) on the approval of a new MA in the centralised procedure when determining an application for a UKMA(GB). Following implementation of the Northern Ireland Protocol, centrally authorised EU MAs continue to apply in Northern Ireland. The MHRA can grant three types of MA: a UKMA(NI) valid only in Northern Ireland or a UKMA(UK) valid throughout the UK, both of which comply with EU law, or a UKMA(GB) which applies to Great Britain only and complies with UK law. The MHRA can use the decentralised or mutual recognition procedure to grant UK MAs (UK) or (GB) based on decisions from EU/EEA countries.
- Also applies in the UK under terms of withdrawal agreement. The data exclusivity and market protection periods have been transposed into UK law by the Human Medicines Regulation 2012 (2012/1916). The date for calculation of these periods in Great Britain (outside of Northern Ireland) will be set by the UKMA.
- Unless there is orphan drug designation, which is treated under a different regulatory data protection regime (see our related briefing for more information)
- Adapted from slide 5 of Data exclusivity, market protection, orphan and paediatric rewards, EMA.
- If the new therapeutic indication with significant clinical benefit relates to a paediatric population, then the applicant will need to choose between (a) an additional six months of SPC term under the Paediatric Regulation, and (b) an extra year of market protection. For further information on paediatric medicines in the EU/EEA and UK, please see our separate briefing on this topic.
- See, Article 6(1) of Directive 2001/83/EC.
- See, for example, European Commission’s “Notice to Applicants”, Volume 2A, Chapter 1, T-472/12 (Novartis Europharm v European Commission), and T-611/18 (Pharmaceutical Works Polpharma v EMA).