
Review of EPO Antibody Decisions in 2023
Last year we provided a review of 19 EPO Board of Appeal decisions relating to “antibody inventions” that were published between January and December 2022. We have now reviewed the decisions published in 2023.
Before discussing these decisions, we should first briefly mention G 2/21, which is a decision by the Enlarged Board of Appeal published in March 2023. G 2/21 considered whether post-published evidence could be used to prove the existence of a technical effect when assessing inventive step. The decision confirms that post-published evidence can in principle be used, but only when the skilled person, with the common general knowledge in mind, and based on the application as originally filed, would derive the technical effect as being “encompassed by the technical teaching” of the originally disclosed invention.
Post-published evidence can of course be important for any invention, but the decision in G 2/21 is particularly relevant for medical use claims. It confirms that post-published data can be relied upon as evidence of a therapeutic effect when considering the inventive step of such claims, but only if it is credible from the application as filed that said effect would actually be achieved. It remains to be seen whether this “credibility threshold” will be assessed more or less strictly than previous approaches to this question, which required an effect to be “plausible”. In any case, the impact of G 2/21 on claims to therapeutic uses of antibodies may be more significant than for other pharmaceuticals, because it is still relatively common for antibody patent applications to be filed with no (or only minimal) direct evidence of a therapeutic effect. As such, we expect to see a steady flow of Board of Appeal decisions in the coming years in which the outcome turns on the ability of the proprietor to rely on post-published evidence for the therapeutic effect of an antibody.
Turning back to Board of Appeal decisions published in 2023 that relate specifically to “antibody inventions”, we identified 18 such decisions. We do not believe that any of these decisions represents a significant shift in EPO practice, nor that any of them will compel changes in the way that examination is conducted.
In our view it remains the case that antibody inventions are not treated differently at the EPO to any other forms of technology. In decisions where “antibody-specific” considerations may be applied, the EPO has done this consistently and in line with the (now largely established) basic principles that are further explored in our advanced guide to drafting and prosecuting antibody inventions at the EPO. The majority of the 2023 decisions support the current practices of the Examining Divisions that we observe in routine prosecution, and which were partly codified in a dedicated section of the EPO’s Guidelines for Examination (G-II, 5.6; first issued March 2021 and revised subsequently – also discussed in our advanced guide).
On a day to day basis the Guidelines may remain a more important source of guidance than the case law, but the 18 decisions covered in this review nonetheless provide confirmation and consolidation of EPO practice, represent useful examples of fact patterns for future reference, and may also help to illustrate developing trends. One of these trends is the increasing number of cases relating to “downstream” developments of existing antibodies, particularly for treating subgroups of patients and specific dosing regimens.
Interestingly, we have continued to see evidence over the year that the EPO is willing to allow broad antibody claims under certain circumstances, for example where the antibody is defined by its epitope. This type of definition is narrower than a claim directed broadly to an “anti-target” antibody, but it provides greater coverage than claims which define the antigen binding domains or complementarity-determining regions (CDRs) by their specific amino acid sequence (“2nd generation” antibody claims). As such, claiming an antibody by its epitope sequence is becoming more popular with applicants looking to capture some additional breadth for antibody variants. This will be encouraging for applicants who may have been forced to accept relatively narrow, structurally-defined antibody claims in the USA and are looking to obtain broader coverage in Europe. We have seen a number of decisions in 2023 which relate to epitope-defined antibodies and we anticipate more Board of Appeal decisions on this point in the future.
Below we provide some statistics from the 18 decisions and discuss some of the more interesting cases in detail.
Statistics
We identified 37 decisions published between 31 January 2023 and 31 December 2023 and which refer to the term “antibody”. 19 of these decisions were not directly concerned with antibody inventions as such and so are not considered further in this review. Of the remaining 18 decisions, 15 concerned appeals from Opposition Division decisions, and 3 concerned appeals from Examining Division decisions. Board 3.3.04 retained its position as the most prolific “antibody Board” with 11 of the 18 decisions (previously 16/19 in 2022). 5 decisions were issued by Board 3.3.08 (previously 2/18 in 2022), and 2 by Board 3.3.07 (previously none in 2022). We discerned no obvious differences in practice between the various Boards.
A breakdown of the key grounds for each decision is provided in Figure 1.
Figure 1
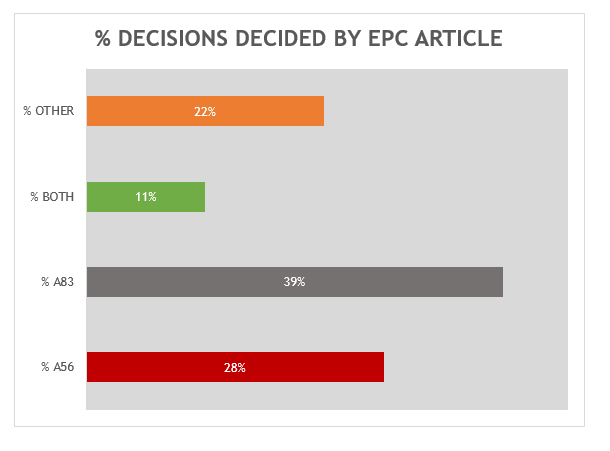
Repeating the trend from previous years, the majority of the 18 decisions were decided primarily on the basis of inventive step (Article 56 EPC) or sufficiency (Article 83 EPC), or both inventive step and sufficiency to some degree. It remains the case that there is often considerable overlap between Article 56 EPC and Article 83 EPC for antibody inventions, and we found the EPO’s assessment of both Articles to be consistent across all of the relevant decisions.
Outside of Article 56 EPC and Article 83 EPC, we found four decisions which were decided on other grounds. Three of these were decided based on novelty (Article 54 EPC). In the remaining decision (T 0699/19) novelty was a key ground, but clarity (Article 84 EPC) was also considered. This case related to an anti-NGF antagonist antibody for use in treating osteoarthritis pain. During the Opposition proceedings, the proprietor introduced amendments to specify that the antibody binds to the same epitope as a reference antibody. These amendments were rejected by the Opposition Division for lacking clarity, since the reference antibody was not structurally defined in the application as filed, nor in the prior art referenced therein. The Board then concluded that the claims lacked novelty, because the prior art disclosed an anti-NGF antagonist antibody for treating osteoarthritis pain in a manner sufficiently clear and complete for it to be carried out by the skilled person. In view of the lack of novelty, the proprietor was forced to narrow the claims to the specific CDR sequences of their antibody. There are no other new “antibody-specific” issues illustrated by this case. However, it serves as a useful reminder that claiming an antibody by reciting a functional feature of a reference antibody may be allowable, but only in circumstances when the reference antibody is itself fully defined.
The 18 decisions can also be divided into four general categories of invention as shown in Figure 2.
Figure 2
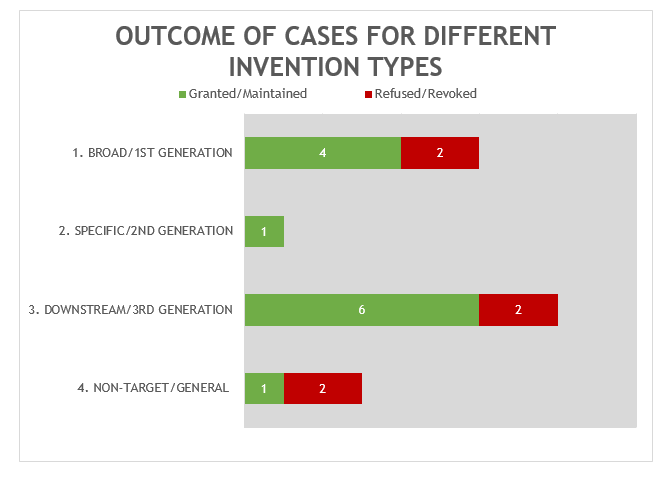
Following a trend that we anticipated last year, the largest category in 2023 (8 decisions – 6 granted/maintained, 2 revoked/refused) is concerned with what we define as “downstream” antibody inventions. These inventions relate to advances which typically arise during downstream development of a pharmaceutical, such as combinations with other therapeutic agents, identification of alternative medical uses, subgroups of patients, dosing regimens and partner diagnostics. In many respects these are not specifically “antibody inventions” because the target binding properties of the antibodies concerned have usually already been established in the art. We find that such inventions are examined by the EPO in the same way as comparable inventions for non-antibody pharmaceuticals. We identified no particular trends in the spread of positive and negative decisions for the applicant/patentee in this category in 2023. It will be interesting to see if G 2/21 has an impact in this respect given that, as noted above, it is still relatively common for applicants to file on antibody inventions with minimal direct evidence of a therapeutic effect.
The next largest category (6 decisions – 4 granted/maintained, 2 revoked/refused) is concerned with what we define as “broad” anti-target antibody inventions, also known as a “1st generation” antibody case. Such cases may entirely lack disclosure of any individual antibody molecule that binds to the target, especially if the invention relates to the identification of the target and/or a link to a particular indication, rather than to an individual antibody or antibodies. In this category we have also included antibodies which are defined by a specific epitope sequence. Although these claims are narrower than broad, anti-target antibody claims, we think they belong in this category because they are nonetheless much broader than the specific “2nd generation” category, which define individual antibodies, typically by their set of CDR or variable chain sequences.
The next category (3 decisions – 1 granted/maintained, 2 revoked/refused) is concerned with antibody inventions that we define as “non-target” or “general” in nature. These inventions are not typically subject to any “antibody-specific” considerations. Rather they are examined in broadly the same way as any other technology at the EPO. Also similar to the above, we do not see any particular trends in the spread of positive and negative decisions for the applicant/patentee in this category, but it is pleasing to see that general antibody platforms, such as antibody libraries for optimising biological characteristics, remain patentable at the EPO.
The final category (1 decision – 1 granted/maintained) is concerned with what we define as “specific” or “2nd generation” antibody inventions. Typically, the invention relates to an individual antibody or antibodies, defined by sequence, and directed to a well-characterised target for which prior art antibodies (often therapeutic antibodies) are known. Readers are likely familiar with the EPO’s approach to such inventions. All antibodies specific for a target are obvious by comparison to prior art antibodies for the same target, unless an unexpected effect is plausibly demonstrated. This can be particularly challenging when the prior art includes high-affinity therapeutic antibodies to the same target. Consistent with the trend from last year, the decision in this category featured Article 56 EPC (inventive step) as a key ground. As mentioned last year, despite the large number of “specific” antibody inventions prosecuted at the EPO, there tend not to be many Board of Appeal decisions relating to this category. This may be because the relatively narrow scope of the claims is less likely to be subject to opposition.
Broad/1st Generation Decisions
T 1624/21 sought to broadly claim an antibody which is capable of specifically binding to a defined epitope of the human 14-3-3 η protein. The main issue in this case was whether the examining division had applied the correct standard of proof when alleging that the claim lacked novelty. The Board of Appeal confirmed that the correct standard of proof to assess whether a specific statement of fact is true or not was the balance of probabilities. “Absolute certainty” is not required. The epitope defined in the claim was not explicitly identified in the prior art and therefore the Board concluded that the Examining Division had applied the incorrect standard of proof. The broad claim was thus maintained.
T 1875/19 related to a bispecific single chain antibody with a first binding domain recognising a specific epitope of human and non-chimpanzee primate CD3 epsilon and a second binding domain recognising a cell surface antigen. The main issue was whether the application sufficiently disclosed that the claimed bispecific antibody could actually recognise the particular epitope of CD3 epsilon. In the Examples of the patent application, a “peptide spotting” assay used to determine antibody-epitope binding demonstrated that some peptides comprising the claimed epitope would not bind the anti-CD3 domain. The Board therefore concluded that there were serious doubts as to whether the bispecific antibody would bind the epitope as defined in the claim, and the claim was revoked in view of insufficient disclosure.
T 0435/20 is interesting in that, despite the EPO being willing in principle to allow claims defining a genus of antibodies by their recognition of a novel epitope, this decision shows that they will also refuse such claims under certain circumstances. The claims here were directed to an antibody defined by binding to a particular epitope of IL-23. The production of antibodies to linear epitopes is often considered routine by EPO, and therefore antibodies directed to such targets can be claimed in functional terms. However, in the present decision, the epitope was a group of co-located residues, not arranged linearly but brought into proximity to each other by protein structure. Such discontinuous or “conformational” epitopes are dependent on secondary structure rather than primary sequence, and hence it is viewed as more challenging to produce antibodies that bind to them. The claims were held to be insufficient and the patent was revoked.
T 1708/18 is another decision which goes against the trend of allowable epitope-defined antibody claims. The claim at issue here related to an isolated antibody that binds to specific epitopes of PCSK9 protein. The Board considered arguments that known anti-PCSK9 antibodies would inherently bind to the epitopes specified in the claim. Using the appropriate standard of proof for assessing whether a statement of fact is true or not (“balance of probabilities”), the Board held that an anti-PCSK9 antibody disclosed in the cited prior art would also bind to the same epitope. The claim therefore lacked novelty and the patent was revoked.
T 0835/21 concerned an antibody that specifically binds to a defined epitope of human LRP6 and exhibits certain additional functional effects. The Board first considered whether the claim to the antibody was sufficiently disclosed. Although the application only disclosed two exemplary antibodies having the features of the claimed invention, the Board concluded that given these examples and the common general knowledge, other examples could be produced. The general view at the EPO is that it is routine to make antibodies to a known target and to screen them for particular functional effects, albeit this could be potentially very time-consuming and tedious. Tedium is not viewed as an undue burden on the skilled person, and so the claims were held to be sufficient.
Perhaps more interesting in this case was the assessment of inventive step, which turned on the additional functional limitation that the antibody is capable of antagonising the Wnt signalling pathway and inhibits Wnt3 and Wnt3a specific signalling activity. It appears that the epitope alone would not have been enough to support an inventive step, because binding to the epitope did not necessarily result in any unexpected technical effect relative to known LRP6 antibodies. Accordingly the inclusion of an additional functional feature was essential to the patentee’s case. The Opposition Division had construed “Wnt3 and Wnt3a specific” to cover antibodies that could also inhibit signalling activity of other Wnt ligands and hence there was no unexpected technical effect relative to known antibodies. The Board disagreed, concluding that the feature implies preferential inhibition of Wnt3/Wnt3a signalling compared to other Wnt ligands, i.e. the antibody does not exclusively inhibit Wnt3/3a signalling, but cannot inhibit signalling of other Wnt ligands to the same degree. The availability of antibodies with this specificity of function was not considered to be obvious from the prior art, and hence the claims were held to be inventive.
T 1478/18 concerned a particular antibody preparation suitable for intravenous administration in humans. The Board considered that the claims were inventive because there was a clear and measurable technical difference between the claimed preparation and that of the prior art. This is broadly consistent with EPO practice relating to other types of pharmaceutical invention.
T 2347/19 related to the use of dexamethasone to prevent neurological adverse events (NAEs) caused by administration of an anti-CD3 antibody. The main issue was whether the anti-CD3 antibody was sufficiently disclosed, because the experiments in the Examples all involved the bispecific anti-CD19xCD3 antibody blinatumomab. Accordingly, the Board considered whether blinatumomab could be generalised to any anti-CD3 antibody. After reviewing the data, the Board held that neurologically adverse events (NAE) could not be unequivocally attributed to the fact that blinatumomab binds CD3, so there would be no rationale for the skilled person to conclude that dexamethasone could prevent NAEs caused by any anti-CD3 antibody. To address this lack of sufficiency, the proprietor limited the claims to an anti-CD19xCD3 antibody.
Downstream/3rd Generation Decisions
T 3165/19 concerned an anti-PCSK9 antibody for use in a method of reducing cardiovascular risk in a high cardiovascular risk patient sub-group, at a specifically defined dose. The closest prior art described an ongoing Phase 3 trial to evaluate the effect of alirocumab (anti-PCSK9) on the occurrence of cardiovascular events in the same patient sub-group. However, no results of the trial had been disclosed. Other prior art documents showed that in the same patient group there was no clear link between reducing LDL-C levels (e.g. with an anti-PCSK9 antibody) and reduced cardiovascular risk. Therefore, the Board concluded that the skilled person would not have expected the trial protocol to be successful in reducing cardiovascular risk in treated patients. Hence, the claimed subject matter was deemed inventive. This is an interesting decision because clinical trial protocols are usually interpreted by the EPO as suggesting a reasonable expectation of success. This is a good example of a case where the patentee was able to argue that doubts remained as to whether following the treatment arm in the clinical trial would lead to effective treatment.
The claim under issue in T 1992/21 was directed to natalizumab for use in treating an inflammatory or autoimmune disease in sub-group of patients, as identified by a blood test for the absence of antibodies against JC virus. The claim also specifies that the treatment is discontinued if indicators for progressive multifocal leukoencephalopathy (PML) are identified in the patient. The closest prior art is a review of three cases of PML that occurred during clinical trials involving natalizumab for treating multiple sclerosis or Crohn’s disease. The claimed invention differed in respect of the blood test for anti-JC virus antibodies, which resulted in a safer treatment. The presence of anti-JC virus antibodies indicates that a previous (“latent”) infection with JC virus has occurred in that patient. The link between latent JC virus infection and increased risk of PML was part of the common general knowledge, and so the skilled person would have recognised that the safest way to treat patients with natalizumab is to treat only those who are not at predisposed to PML i.e. those who do not possess latent JC virus infection. Accordingly, the claims were held to lack an inventive step and the patent was revoked.
T 0654/20 concerned a composition comprising an antibody that selectively binds to c-kit and interferes with c-kit signalling for use in a method of stem cell engraftment in SCID patients, involving a series of method steps involving the patient. The arguments raised by the opponent related to a lack of sufficiency, based on the fact that experiments disclosed in the patent application were only carried out on mice. The Board of Appeal did not agree, because the claimed method relied on an underlying mechanism of c-kit signalling that was shared between mice and humans. Accordingly, the experimental results could credibly be extrapolated from mouse to human and the claims were held to be sufficiently disclosed.
T 1394/21 related to a VISTA antagonist antibody and a PD-1 antagonist antibody for use in treating cancer. The Opposition Division had previous concluded that identifying antagonistic anti-VISTA antibodies represented an undue burden, because there was no enabling disclosure of specific anti-VISTA antibodies in the application as filed. However, the Board disagreed with this conclusion. The application provided the required information for producing anti-VISTA antibodies in general as well as assays for identifying other antagonistic antibodies. Therefore, the skilled person would be able to provide antagonistic anti-VISTA antibodies based on the teaching in the application as filed. Separately, it was discussed whether the application as filed rendered the claimed therapeutic effect credible. In this case, the negative results identified by the respondent could not be assessed are there were no controls. Instead, the Board found that it was credible and predictable for the skilled person that the claimed antibody combination was suitable for treating cancer. The claims were therefore held to be sufficiently disclosed.
The claim under issue in T 1675/20 related to a pharmaceutical combination for use in medical treatment which comprised dendritic cells associated with a target antigen and a second “boost” composition comprising the target antigen in soluble form and a co-stimulatory antibody directed to one of a number of T-cell markers. In the application as filed, only one specific combination was tested for its therapeutic effect in a murine model of cancer. Immune responses were also measured in response to some other boost compositions. Accordingly, the issue considered by the Board was whether the application nevertheless renders it plausible that all of the claimed combinations could achieve the therapeutic effect. After reviewing the available data, the Board were not convinced that the application shows an increase in immune response for combinations having an anti-ICOS antibody as the co-stimulatory antibody. Therefore, the claims were not sufficiently disclosed across their entire scope and the patent was revoked.
T 0885/21 sought to claim conjugates of an antibody that specifically binds a cancer antigen with a cytotoxin, for use as a medicament. The main contentious issue was sufficiency of disclosure, in particular the appellant presented arguments that the claims would cover conjugates unsuitable for the medical use and examples of antibody-cytotoxin conjugates that failed in clinical trials due to toxicity or lack of efficacy. The Board firstly noted that the claims are purpose-limited and so would functionally exclude conjugates which are unsuitable for use in therapy. The Board then reviewed the examples of the application as filed and concluded that the skilled person would have sufficient guidance to carry out the invention, absent evidence to the contrary. The proprietor then relied on post-published experimental results to confirm that the claimed conjugates exhibit optimised characteristics, in order to establish inventive step. Significantly, the Board considered that effects described in the post-published evidence were encompassed by the technical teaching of the application and may therefore be relied upon for inventive step, in accordance with the principles confirmed in G 2/21.
Non-target/General Decisions
T 2455/19 related to composition comprising (i) an antibody fragment (“immunobinder”) that contained solubility enhancing motifs at defined positions in the heavy chain and (ii) a pharmaceutically acceptable carrier. The closest prior art disclosed the same immunobinder so the only difference was the presence of the pharmaceutically acceptable carrier. The Opposition Division decided that the claims were inventive, because the presence of the carrier implied a therapeutic purpose, and there was no hint that the prior art fragment was useful in therapy. The Board of Appeal disagreed, because they considered that the prior art included compounds which could be interpreted as pharmaceutically acceptable carriers. Therefore, there was no difference between the claim and the prior art, and the claim was revoked.
T 0416/20 concerned a set of polypeptides which associated in the presence of a cell expressing two antigens to form a structure that corresponds to a bispecific antibody. The key feature of the claims was that the polypeptides did not associate with each other in the absence of a cell that has both antigens on the cell surface. The Board construed the term “not associated” to include polypeptides that may associate weakly, when taken in context of the patent application as filed. In view of this claim construction, the prior art was held to disclose an identical set of polypeptides, namely VL and VH domains that form a bispecific antibody. The claims therefore lacked novelty and the patent was revoked.
The claims of T 0047/22 related to a method of designing an antibody library for optimisation of a biological property. The Board found it credible that the method would work and furthermore found that there was no teaching towards the key features (derivation from same germline sequence by somatic hypermutation) in the prior art. The claims were thus held to be sufficiently disclosed and inventive. It interesting to note that methods of optimising antibody properties (and therefore antibodies arising from such methods) can be patented. However, the conclusion reached by the Board based on the facts in this case is broadly consistent with EPO practice relating to other types of inventions.
Specific/2nd Generation Decisions
T 1669/19 was an appeal by the opponent against a decision by the Opposition Division to uphold the patent in amended form. The Opposition Division concluded that the claims were entitled to claim priority, which excluded an otherwise relevant document from the prior art. The appellant argued that the claims were not entitled to the priority date. In particular, one issue was that the priority application refers to “fully” human antibodies, whereas the claims simply refer to human antibody. The Board rejected this argument as it was not proven that a genuine distinction could be made between human and “fully” human antibodies. The appellant then ran an inventive step attack based on a document relating to an antibody with a different sequence. In view of the improved potency of the claimed anti-human PCSK9 antibodies, the Board acknowledged that the claims involved an inventive step.
Trends and Conclusions
As observed last year, there appears to be a continued lack of interest in opposing patents in the specific/2nd generation anti-target category. This explains why there is only 1 decision in this category in the present review, despite this being the most common type of antibody claim that we observe in prosecution.
This year we have also seen an increasing number of decisions relating to downstream developments of a pre-existing antibody. This is different to last year, when we identified that more decisions were concerned with the “target binding” properties of antibodies. We suggest that there is a continued interest in protecting such downstream developments, given that many anti-target antibody therapeutics are now well-established. For example, we expect to see more cases relating to new medical uses, combinations of antibodies with other compounds, patient sub-groups and dosage regimens.
Notwithstanding the above, the Board of Appeal decisions from 2023 confirm that it is still possible to obtain broad antibody claims in some circumstances, such as when the antibody is defined by its epitope. We therefore expect decisions relating to epitope-defined antibodies to be more common in the future. As anticipated last year, we also expect to see more decisions which define a class of antibodies as “competing for binding” with a reference antibody.
As a practice point, when drafting new patent applications, we recommend including all available sequence information and experimental data relating to the antibody, including in particular any experiments which demonstrate interaction of the antibody with its target epitope, any competition binding studies performed with other reference antibodies, any information relating to the epitope bound by the antibody, and any functional assays which may be used to demonstrate (or at least imply) a likely therapeutic effect.
J A Kemp has extensive expertise in handling patents relating to antibodies. For more information, see our Antibodies and Biologics specialism.